Neue Studie: 14,9% Unterschied zwischen Mensch und Schimpanse
Seit 2005 das Schimpansengenom erstmals vollständig entschlüsselt wurde, wird fortwährend verbreitet Mensch und Schimpanse seien genetisch zu 98-99% identisch. Eine weitere Studie widerlegt nun zum wiederholten Male diesen Mythos. Yoo et al. (2025) sequenzierten vollständig die Genome vom gemeinen Schimpansen, westlichen Gorilla, Borneo- und Sumatra-Orang-Utan sowie dem Siamang (ein großer Gibbon) und verglichen diese vollständig miteinander. Das ist besonders, denn vorherige Studien taten dies nicht immer.
Lukas Kuderna (2025) kommentierte die Studie dazu:
„Kurz nachdem die erste menschliche Genomsequenz 2003 fertiggestellt worden war, wurde eine Schimpansen-Sequenz veröffentlicht. Es folgten Sequenzen für andere Großaffen wie Gorillas, Sumatra-Orang-Utans und Bonobos und für kleine Affen, die weniger eng mit dem Menschen verwandt sind als Großaffen.
Diese Genome boten eine wertvolle Gelegenheit, die genetischen Unterschiede zu katalogisieren, die sich im Laufe der Evolution der Menschenaffen angesammelt haben, einschließlich der Veränderungen, die nur beim Menschen auftreten. Da es sich bei diesen ersten Veröffentlichungen jedoch um unvollständige Entwürfe handelte, konnten nur Vergleiche zwischen ordnungsgemäß aufgelösten Teilen des Genoms angestellt werden.
Diese Studien konzentrierten sich daher nur auf relativ kleine Unterschiede und schlossen extrem sich wiederholende Sequenzen und großräumige strukturelle Unterschiede, wie Inversionen und Duplikationen von Genomsequenzen, aus.„ [Hervorhebung hinzugefügt.]
Yoo et al. (2025) bezogen nun auch die Sequenzen ein, die als Indelmutationen (Einschübe und Auslassungen mehrerer Basen), Duplikationen oder Inversionen (umgedrehte Sequenzen) gedeutet werden und deshalb keinen direkten Base-für-Base-Vergleich erlauben.
Einfach ausgedrückt legten sie einfach je 2 Genome vollständig nebeneinander und sahen sich das Gesamtbild an. Das führte sie zu einer sogenannten „Lücken-Divergenz“. Sie bezeichnet den Anteil in Prozent der beiden Genome, der durch die genannten strukturellen Unterschiede nicht direkt vergleichbar ist.
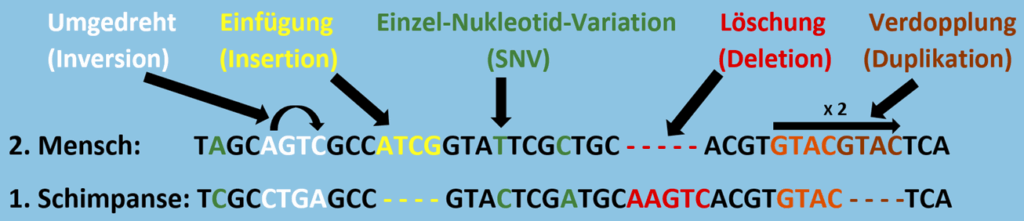
Bei Mensch und Schimpanse stellten die Wissenschaftler eine eine Lücken-Divergenz von 12,5%, bzw. 13.3% fest. – Je nachdem ob man Mensch mit Schimpanse vergleicht oder Schimpanse mit Mensch (Bezogen auf die Autosomen, Unterschiede in den Geschlechtschromosomen werden hier der Einfachheit halber ausgelassen). Diese Ergebnisse finden sich allerdings nicht direkt in der Studie, sondern in ihren ergänzenden Daten. Damit ist die oft zitierte Behauptung Mensch und Schimpanse seien genetisch zu 98-99% identisch, faktisch erneut widerlegt.
Der Schöpfungswissenschaftler Casey Luskin hat einige Artikel zu dieser Studie und damit zusammenhängenden Diskussionen verfasst. Er schreibt dazu:
„Und es [das Schimpansengenom] zeigt eine „“Lücken-Divergenz“ – d. h. einen Unterschied – von 12,5 Prozent zum menschlichen Genom! Und wenn du dir die „Lücken-Divergenz“ ansiehst, bei der das menschliche Genom das Ziel und das Schimpansengenom die Abfrage ist, beträgt der Unterschied 13,3 Prozent. Lass mich deutlich sein:
Laut dieser Studie sind das menschliche und das Schimpansengenom nicht zu 98,8 Prozent identisch (oder zu 1,2 Prozent unterschiedlich) […]. Tatsächlich sind sie zu nicht mehr als 87,5 Prozent ähnlich – d. h., das menschliche und das Schimpansengenom unterscheiden sich um mindestens 12,5 Prozent, wenn nicht sogar um 13,3 Prozent! Tatsächlich ist der Unterschied von 13,3 Prozent relevanter, da er widerspiegelt, wie ähnlich das gesamte menschliche Genom dem Schimpansengenom ist.“ [Hervorhebung hinzugefügt]
Diese 13,3% beziehen sich wie bereits erwähnt nur auf die Sequenzen, die nicht durch einen direkten Base-für-Base-Vergleich vergleichbar sind. Die restlichen 86,7% unterscheiden sich nur in einzelnen Basen (im Englischen als „SNV“ abgekürzt) und hier unterscheiden sich die Sequenzen des Menschen nach der Studie um 1,6% von denen des Schimpansen.
Damit ergibt sich ein Gesamtunterschied von 14,9% (auch aufgerundet als 15% zitiert). Bei den Ergebnissen verwundert es nicht, dass die Studie bereits von schöpfungswissenschaftlicher Seite aufgegriffen wurde (siehe Luskin und Borger & Scholl im Studium Integrale Journal, 32. Jahrgang, Heft 2). Dem folgte natürlich Kritik von naturalistischer Seite, bis hin zu Vorwürfen von mutwilliger Grafikmanipulation:
Naturalistische Kritik:
1) Grafikmanipulation: In den ergänzenden Daten der Studie findet sich eine Grafik, die die Ergebnisse der einzelnen Vergleiche darstellt. Sie enthält sowohl die Unterschiede zwischen Mensch und den einzelnen Affenarten, als auch innerartliche Variationen.
Luskin verwendete nun diese Grafik für einen seiner Artikel, schnitt aber die innerartlichen Variationen raus. Das führte zu Kritik. Beispielsweise schreibt der Evolutionsbiologe Martin Neukamm:
„Das Aufdecken dieser Taktik ist aus zwei Gründen aufschlussreich: Erstens entzieht die vollständige Darstellung der Daten der Argumentation der Kreationisten jede Grundlage. Liegen die genetischen Unterschiede bei Schimpansen bereits bei 8,8 % und bei Gorillas bei 13,8 %, erscheinen 13,3 % zwischen Mensch und Schimpanse sehr gering. LUSKIN konnte seine Argumente nur platzieren, indem er genau diese zentrale Information verschwieg. Oder möchte das Discovery Institute bestreiten, dass alle Gorillas einen gemeinsamen Vorfahren haben?“
Luskin reagierte, indem er die vollständige Grafik in seinen Artikel einfügte und mit folgender Anmerkung versah:
„Anmerkung des Autors (8. Juli 2025): Ich veröffentliche hier die vollständige Grafik aus Abbildung III.12 der ergänzenden Daten von Yoo et al. (2025), da einige Leute behaupten, ich hätte ursprünglich Daten aus der Grafik weggelassen, weil sie irgendwie meine Argumentation widerlegt hätten.
Nein, das ist falsch. Der einzige Grund, warum ich nur die Genomvergleiche zwischen Mensch und Affe (die oberen acht Kurven) veröffentlicht habe, war der der Vereinfachung, da dies der einzige Teil der Grafik war, der für meine Argumentation relevant war; die intraspezifischen genetischen Unterschiede (die unteren fünf Kurven) sind irrelevant.
Meine Argumentation lautet nicht, dass genetische Unterschiede zwischen Mensch und Affe notwendigerweise die Evolution widerlegen, sondern dass Evolutionisten, die jahrzehntelang behauptet haben, Menschen seien genetisch nur zu 1 Prozent von Affen verschieden, falsch lagen. Die oberen acht Kurven veranschaulichen meinen Standpunkt sehr deutlich.
Es gab und gibt keine Daten in der Grafik, die meine Argumentation negativ beeinflusst hätten, und ich habe immer auf die ergänzenden Daten verwiesen, wo Interessierte den vollständigen Datensatz einsehen können. Hier gibt es nichts zu verbergen. Tatsächlich bin ich bereits ausführlich auf Einwände bezüglich der intraspezifischen genetischen Unterschiede eingegangen, bevor die aktuellen Kritiker mit ihren Kommentaren begannen.“ [Hervorhebung hinzugefügt]
Damit kann dieser Vorwurf als haltlos und irreführend zurückgewiesen werden.
2) Hohe innerartliche Unterschiede relativieren den Unterschied zwischen Mensch und Schimpanse: Hierzu nochmal Neukamm:
„Zwischen Schimpansen und ihren nächsten Verwandten, den Zwergschimpansen (Bonobos), deren DNA zu 99,1 % identisch ist, ist die Gap divergence mit 11,7 % ähnlich hoch. Dass beide Arten trotzdem nächstverwandte Spezies mit einem gemeinsamen Vorfahren sind, bestreiten nicht einmal die Kreationisten. Vor diesem Hintergrund ist eine Gesamtdifferenz von 15 % zwischen Mensch und Schimpanse, deren Entwicklungslinien sich vor 7 Mio. Jahren trennten, evolutiv hochplausibel. Am Primaten-Stammbaum hat sich nichts geändert. […] Liegen die genetischen Unterschiede bei Schimpansen bereits bei 8,8 % und bei Gorillas bei 13,8 %, erscheinen 13,3 % zwischen Mensch und Schimpanse sehr gering.“
Damit weichen Kritiker dem primären Argument aus. Denn wie Luskin in obiger Anmerkung bereits klarstellte, ging es ihm nicht primär darum eine evolutionäre Verwandschaft von Mensch und Schimpanse zu widerlegen. Er stellte lediglich klar, dass sie sich genetisch um deutlich mehr als 1-2% unterscheiden.
Denn über Jahre wurde verbreitet das ganze Genom von Mensch und Schimpanse sei zu 98-99% identisch und dies sei ein Beleg ihrer gemeinsamen Abstammung. Einige Beispiele:
Der ehemalige Wissenschaftskommunikator Bill Nye schrieb 2014 in seinem Buch „Undeniable„:
„Mit zunehmendem Verständnis der DNA haben wir erkannt, dass wir etwa 98,8 Prozent unserer Gensequenz mit Schimpansen gemeinsam haben. Dies ist ein eindrucksvoller Beleg dafür, dass Schimpansen und Menschen einen gemeinsamen Vorfahren haben.“
„Wir teilen mehr als 98 Prozent unserer DNA und fast alle unsere Gene mit unserem nächsten lebenden Verwandten, dem Schimpansen.“
Kevin Williamson schrieb 2016 im National Review:
„Etwa 99 Prozent unserer DNA ist identisch mit der von Schimpansen.“
Und im Scientific American war 2004 zu lesen:
„Die meisten Studien zeigen, dass beim Vergleich der Genomregionen von Schimpansen und Menschen eine Sequenzidentität von etwa 98,5 Prozent besteht.“
Viele weitere solcher Bsp. könnte man anführen. Auf die (erneute) eindeutige Widerlegung solcher Falschaussagen mit Ausweichen und Relativierung zu reagieren, ist alles andere als aufrichtig. Wissenschaftlich korrekt und redlich wäre es gewesen, die Widerlegung des bisherigen Argumentes einzugestehen und dann ggf. begründet darzulegen, warum die neue Datenlage der gemeinsamen Abstammung trotzdem nicht widerspricht.
So aber entsteht ein Strohmann-Argument – zumindest in Bezug auf die Kritik auf Luskin: Man schafft mit innerartlichen Unterschieden einen relativierenden Vergleich, um zu widerlegen, dass der höhere Unterschied gemeinsamer Abstammung widerspricht. – Dabei hat Luskin dies nie behauptet.
Darüber hinaus ist die Gleichsetzung von zwischenartlichen Unterschieden mit innerartlichen Unterschieden kategorisch falsch. Verschiedene Arten unterscheiden sich neben Einzel-Nukleotid-Unterschieden in gemeinsamen Genen vor allem in unterschiedlichen Genen und zusätzlichen, bzw. fehlenden Genen. Individuen derselben Art hingegen haben die gleichen Gene, welche sich lediglich in den Allelen unterscheiden. Innerartliche Unterschiede betreffen nur Variationen innerhalb der gleichen Gene, zwischenartliche Unterschiede betreffen darüber hinaus verschiedene und andersartige Gene. Wer versucht beides gleichzusetzten, vergleicht sozusagen Äpfel mit Birnen.
Borger & Scholl schreiben außerdem zu den innerartlichen Unterschieden:
„Aus Schöpfungsperspektive kann man die genetischen Unterschiede in nicht-codierenden Bereichen auch anders deuten: Sie repräsentieren oft redundante genetische Information, die innerhalb eines Grundtyp Variation ermöglicht, ohne die grundlegende biologische Identität zu zu verändern (Borger 2017). Solche redundanten oder duplizierten Bereiche können verloren gehen oder variieren, ohne essenzielle Funktionen zu beeinträchtigen – und dennoch zur Vielfalt innerhalb einer Art beitragen.
Somit sprechen die Daten für ein flexibles, aber robustes genetisches System, das Vielfalt hervorbringen kann – ganz im Sinne eines durchdachten Designs.“ (Borger P & Scholl B (2025) Der genetische Unterschied zwischen Mensch und Schimpanse beträgt 15%. Studium Integrale Journal, 32. Jahrgang, Heft 2, S. 90.)
Innerartliche Unterschiede wirken sich also weder negativ auf Luskins Argumentation aus, noch machen sie eine gemeinsame Abstammung von genetisch zu knapp 15% unterschiedlichen Arten plausibler. Darüber hinaus sind sie auch ohne weiteres schöpfungstheoretisch deutbar.
3) Die Lücken-Divergenz bestehe überwiegend aus Junk-DNA und könne daher ignoriert werden: Das Argument lautet, die Lücken-Divergenz bestehe größtenteils aus nutzlosen Wiederholungen, die nicht vom gemeinsamen Vorfahren stammen, keinen biologischen Unterschied ausmachen und daher für die Rekonstruktion evolutionärer Verwandtschaftsverhältnisse irrelevant seien.
Das ist eine Wiederbelebung eines alten und längst widerlegten Argumentes. Als man entdeckte, dass nur 2% unserer Gene für Proteine codieren, schloss man einfach, dass die restlichen Gene funktionslose Überreste der Evolution seien:
Richard Dawkins schrieb 1976 in seinem Buch „The Selfish Gene„:
„Der wahre ‚Zweck‘ der DNA ist es, zu überleben, nicht mehr und nicht weniger. Die einfachste Erklärung für die überflüssige DNA ist die Annahme, dass sie ein Parasit oder bestenfalls ein harmloser, aber nutzloser Passagier ist, der in den Überlebensmaschinen mitfährt, die von der anderen DNA geschaffen wurden.“
Und Francis Crick schrieb 1980 in Nature:
„Ein Großteil der DNA in höheren Organismen ist kaum besser als Müll und kann mit der Ausbreitung eines nicht allzu schädlichen Parasiten in seinem Wirt verglichen werden.“
In dem Sinn schreibt auch der theistische Evolutionist und Biologe Joshua Swamidass auf X zu der aktuellen Studie:
„Sehen Sie sich diesen Abschnitt von Chromosom 16 an. Der große rosa Block stimmt nicht mit dem von Schimpansen überein, sondern ist lediglich repetitive Satelliten-DNA beim Menschen. Er trägt also zur gesamten Lücken-Divergenz bei, obwohl es sich um triviale Cut-and-Paste-Wiederholungen handelt. Interessanter und wichtiger ist die Divergenz der blauen und gelben Abschnitte, die „ausgerichtete” Divergenz.“
Wie auch Borger & Scholl feststellen, hat diese Annahme der Junk-DNA der biologischen Forschung über Jahre geschadet, da wichtige Bereiche unseres Genoms lange ignoriert wurden und so unser Verständnis davon, wie unser Genom wirklich funktioniert, verzörgert wurde. Das Argument in seiner allgemeinen Form ist seit langem widerlegt. Inzwischen kennt man die Funktion von dem Großteil unseres Erbgutes oder hat deutliche Hinweise auf diese. Für Details siehe z.B. dieser Artikel.
Auch im konkreten Fall der sich wiederholenden Sequenzen der Lücken-Divergenz ist die Bezeichnung als „nutzlos“ nicht haltbar. Borger & Scholl:
„In diesem dreidimensionalen Aufbau spielen gerade die nicht-codierenden DNA-Sequenzen eine zentrale Rolle: Sie steuern wann, wo und wie stark Gene abgelesen werden (Borger 2017; 2021; ENCODE 2020). Es ist daher völlig plausibel, dass gerade diese nicht-codierenden Abschnitte zur genetischen Variation innerhalb von Arten beitragen – nicht durch Veränderungen an den Genen selbst, sondern durch Alternative Steuerung der Genexpression (Faulkner et al. 2009). Solche Regulationsunterschiede können tiefgreifende Auswirkungen auf Entwicklung, Verhalten oder Anpassungsfähigkeit haben.“
Auch von naturalistischen Wissenschaftlern wurde die Funktionslosigkeit dieser Wiederholungen bereits widerlegt. Im April 2025 erschien in dem Journal Nucleic Acids Research ein Artikel, dessen Autoren zum Teil auch an der Studie von Yoo et al. beteiligt waren. Dort werden besagte Wiederholungen entgegen der Stimmen mancher Kritiker als funktionell beschrieben:
„Nicht-kanonische (Nicht-B-)DNA-Strukturen – z. B. gebogene DNA, Haarnadeln, G-Quadruplexe (G4s), Z-DNA usw. –, die sich an bestimmten Sequenzmotiven (z. B. A-phasigen Wiederholungen, invertierten Wiederholungen usw.) bilden, haben sich als wichtige Regulatoren zellulärer Prozesse und Treiber der Genom-Evolution herausgestellt. Aufgrund ihrer repetitiven Natur und potenziell ungenauen Sequenzen, die mit Short-Read-Technologien erzeugt werden, sind sie jedoch bislang nur unzureichend untersucht worden.
Hier charakterisieren wir solche Motive umfassend in den Long-Read-Telomere-to-Telomere (T2T)-Genomen von Menschen, Bonobos, Schimpansen, Gorillas, Borneo-Orang-Utans, Sumatra-Orang-Utans und Siamangs. Nicht-B-DNA-Motive sind in den genomischen Regionen, die zu T2T-Assemblierungen hinzugefügt wurden, angereichert und nehmen 9 %–15 %, 9 %–11 % bzw. 12 %–38 % der Autosomen und Chromosomen X und Y ein. G4s und Z-DNA sind in Promotoren und Enhancern sowie an Replikationsursprüngen angereichert.
Repetitive Sequenzen enthalten mehr Nicht-B-DNA-Motive als nicht-repetitive Sequenzen, insbesondere in den kurzen Armen akrozentrischer Chromosomen. Die meisten Zentromere und/oder ihre flankierenden Regionen sind mit mindestens einem Nicht-B-DNA-Motivtyp angereichert, was mit einer potenziellen Rolle von Nicht-B-Strukturen bei der Bestimmung von Zentromeren übereinstimmt. Unsere Ergebnisse unterstreichen die ungleiche Verteilung der vorhergesagten Nicht-B-DNA-Strukturen über die Genome von Menschenaffen und deuten auf ihre neuartigen Funktionen in bisher unzugänglichen Genomregionen hin.“ [Hervorhebung hinzugefügt]
Luskin kommentiert dieses Zitat in seiner Antwort auf diesen Kritikpunkt mit:
„Entscheidend ist, dass die Anzahl der Kopien von Wiederholungen dazu beiträgt, die Form dieser Nicht-B-DNA zu bestimmen, was Auswirkungen auf genomische Interaktionen und die dreidimensionale Struktur der Chromosomen im Zellkern haben kann. Wir können also diese DNA-Unterschiede nicht einfach ignorieren, nur weil sie als unterschiedliche Kopienzahlen repetitiver DNA auftreten. Diese unterschiedlichen Kopienzahlen können bedeutende funktionelle Unterschiede mit sich bringen.“ [Hervorhebung hinzugefügt]
Das Argument der Funktions- und Bedeutungslosigkeit der sich wiederholenden Sequenzen in der Lücken-Divergenz richtet sich damit direkt gegen den aktuellen Kenntnisstand der Biologie und ist haltlos.
4) Die großen Unterschiede als technische Fehler: Die meisten Kritiker erkennen die Existenz einer Lückendivergenz von knapp 15% an, sie spielen lediglich ihre Relevanz herunter. Doch es wurde auch die Behauptung aufgeworfen, die Lücken-Divergenz sei nur auf technische Fehler während des Vergleichs zurückzuführen.
In der Studie selbst geht laut Luskin nur eine Stelle in diese Richtung:
„Die Lücken-Divergenz zeigte im Vergleich zu Einzel-Nukleotid-Varianten einen 5-fachen bis 15-fachen Unterschied in der Anzahl der betroffenen Megabasen, was auf sich schnell entwickelnde und strukturell variierende Regionen des Genoms sowie auf technische Einschränkungen bei der Ausrichtung in repetitiven Regionen zurückzuführen war (ergänzende Abbildungen III.11 und III.12).„
In den ergänzenden Daten findet sich noch eine etwas konkretere Stelle:
„Die Lücken-Divergenz ist definiert als der Anteil der Positionen im Ziel-Haplotyp, die nicht mit dem anderen Haplotyp abgeglichen sind, was auf biologische Prozesse (z. B. Genverlust/-gewinn und Insertionen/Deletionen), fehlende Daten oder technische Probleme (z. B. Fehlschläge beim Abgleich aufgrund von SVs, repetitiven Elementen usw.) zurückzuführen sein könnte.“
Wenn man diese Zitate aufmerksam liest, wird deutlich, dass es nicht um Fehler geht, die Menschen oder Geräte im Labor gemacht hätten. Vielmehr werden die technischen Schwierigkeiten durch eben jene strukturellen Unterschiede verursacht, die die Lücken-Divergenz ausmachen! Um es mit den Worten Luskins zu sagen:
„Um es ganz offen zu sagen: Nach ihrer Definition ist ein „technischer Fehler“ nicht gleichbedeutend mit einem Fehler, der im Labor gemacht wurde. Nach ihrer Definition bedeutet technischer Fehler = Ausrichtungsfehler = unterschiedliche DNA. Unabhängig davon, wie wir sie bezeichnen, gibt es echte genetische Unterschiede zwischen Menschen und Schimpansen – tatsächlich etwa 15 Prozent des Genoms – und diese Unterschiede verdienen es, anerkannt zu werden.“
5) Die neuen Affengenome sind unzuverlässig: Ein weiterer Einwand besteht darin, die Zuverlässigkeit der neu sequenzierten Affengenomme anzuzweifeln. Diese Kritik stützt sich auf die Formulierung „fehlende Daten“ in einem der hier zuletzt genannten Zitate. Man könnte das so verstehen, dass während der Sequenzierung etwas schief lief und die entsprechenden Daten deshalb in der Studie nicht verwendet werden konnten.
Doch das ergibt im Kontext keinen Sinn. Das Besondere an der Studie ist ja eben die vollständige Sequenzierung, wie es auch schon im Titel der Studie heißt: „Complete sequencing of ape genomes„. Das wird auch in der Studie von den Autoren wiederholt:
„Wir erreichen Kontiguität auf Chromosomenebene mit hoher Sequenzgenauigkeit (<1 Fehler in 2,7 Megabasen) und sequenzieren 215 lückenlose Chromosomen vollständig von Telomer zu Telomer. Wir entschlüsseln schwierige Regionen wie den Haupthistokompatibilitätskomplex und Immunglobulin-Loci […] Fortschritte in der Long-Read-Sequenzierung und neue Assemblierungsalgorithmen waren erforderlich, um die Herausforderung der Wiederholungen zu bewältigen und die erste vollständige Telomer-zu-Telomer-Assemblierung (T2T) des menschlichen Genoms zu erreichen.
Mit denselben Methoden haben wir kürzlich sechs weitere Paare vollständiger Geschlechtschromosomen aus verschiedenen Zweigen der Affenphylogenie veröffentlicht. Obwohl diese ersten Projekte auf haploide Chromosomen abzielten und eine umfangreiche manuelle Kuratierung erforderten, ermöglichen verbesserte Assemblierungsmethoden nun die vollständige Assemblierung diploider Chromosomen. Mit diesen Methoden präsentieren wir hier vollständige, phasierte, diploide Genome von sechs Affenarten und stellen alle Daten und kuratierten Assemblierungen der wissenschaftlichen Gemeinschaft frei zur Verfügung.“ [Hervorhebung hinzugefügt]
Die für die Studie sequenzierten Genome sind vollständig, auch diese Kritik läuft ins Leere.
Fazit:
Der genetische Unterschied zwischen den vollständigen Genomen von Mensch und Schimpanse beträgt mindestens 14,9%, das haben Yoo et al. zweifelsfrei nachgewiesen. Keiner der genannten Kritikpunkte ist stichhaltig. Es ist bezeichnend und etwas ironisch, dass ausgerechnet die Kritik derer, die Kreationisten und Schöpfungswissenschaftler nur zu gerne als „unwissenschaftlich“ und z.T. Schlimmeres titulieren, wissenschaftlich nicht haltbar ist. Sie besteht aus der Widerlegung nie getätigter Aussagen (sog. „Strohmänner“), Kategoriefehlern, unaufmerksamem Lesen, Ausweichversuchen und veraltetem, längst widerlegtem biologischem „Wissen“.
Über viele Jahre wurde verbreitet unser gesamtes Genom würde sich nur um wenige Prozent von dem des Schimpansen unterscheiden und dies sei ein Beleg gemeinsamer Abstammung. Dieses Argument haben Yoo et al. widerlegt. Ob der Unterschied von 14,9% durch Mutation und Selektion in 6-7 Millionen Jahren zu erklären ist, soll hier nicht diskutiert werden, doch es wäre im Sinn der wissenschaftlichen Ergebnisoffenheit und Redlichkeit nur folgerichtig, wenn Evolutionstheoretiker diese Frage zumindest in Betracht ziehen würden.
Aus schöpfungswissenschaftlicher Sicht ist der genetische Unterschied so oder so nebensächlich. Ähnlichkeiten können hier immer funktionell, bzw. als Handschrift des gemeinsamen Schöpfers verstanden werden. Nur die Evolutionstheorie hat das Problem zu große Unterschiede in zu kurzer Zeit nicht erklären zu können.
Anhang:
Neukamms kritischer Artikel scheint bislang nicht von schöpfungswissenschaftlicher Seite registriert worden zu sein, denn manche seiner Kritikpunkte werden weder in Luskins zahlreichen Artikeln, noch im Artikel von Borger & Scholl behandelt. Deshalb habe ich Dr. Peter Borger eine Mail mit Fragen zu einigen Passagen aus Neukamms Artikel geschrieben. Mit seiner freundlichen Erlaubnis werden nun seine Antworten angehängt:
Dr. Peter Borger: „Neukamm argumentiert an mehreren Stellen voraussetzungsbeladen. Er deutet InDels, Inversionen und Translokationen automatisch als Mutationsprodukte, ohne dass diese Interpretation empirisch zwingend wäre. Versteht man solche strukturellen Unterschiede stattdessen als Teil eines ursprünglich Genoms (das ich als Baranom beschreibe), kollabiert sein Deutungsrahmen an entscheidenden Stellen. Es gäbe einfach die Zeit nicht all diese Mutationen zu selektieren. Zudem ist die Population der Vormenschen zu klein. Die Fixierung 5 solcher Mutationen würden schon um die 8 Millarden Jahren benötigen (Sanford).
Hinzu kommt noch, dass mehrere der von ihm genannten InDel-Ereignisse – besonders im Gehirn – sofort zu völlig neuen Genen oder funktionellen Einheiten führen müssten. Die Lücken werden nämlicht von human-spezifischen Gene besetzt. Das ließe sich nur als eine Art „mutationales Wunder“ deuten, einschließlich nicht-zufälliger Mutationsmechanismen. Aus meiner Sicht bleibt hier letztlich nur mein in Darwin Revisited dargestelltes Evolutionsmodell als konsistente Erklärung.
Auch seine Behandlung der TE-Regionen folgt dem alten Junk-DNA-Paradigma. Dass ausgerechnet Moran (2023) als Referenz dient, ist bezeichnend, denn Moran (und Graur) gehören zu den entschiedensten Vertretern der Junk-DNA-Hypothese. Dieses Paradigma ist jedoch weder experimentell abgesichert noch vereinbar mit dem heutigen Wissen über genomische Regulation. Die pauschale Einstufung von über 90 % der TEs als „funktionslos“ ist aus dieser Perspektive nicht haltbar. Man muss das Genom als 4D Wet-Computer betrachten (Nasse Computer), wobei die lineare DNA sich zu 3D funktionalität faltet und sich ständig verändert, weil immer wieder neue Programme aus- und eingeschaltet werden müssen (Veränderung in der Zeit = 4D).
Was Graur, Moran und auch Neukamm übersehen, ist, dass Genome nicht linear funktionieren, sondern in einer vierdimensionalen Architektur operieren. Die von ihnen als „junk“ abgewerteten Elemente tragen wesentlich zur räumlichen Struktur des Genoms bei – insbesondere LINEs und SINEs. Sie operieren in diesen dynamische Chromatin-regulierende Prozessen. Sie wirken nicht nur als regulatorische Schalter, sondern auch als strukturelle Bausteine für LADs, TADs und Isochore. Damit bestimmen sie, wo regulatorische Plattformen entstehen – unterschiedlich in den rund 400 differenzierten menschlichen Zelltypen (obwohl alle aus einer einzigen Zygote hervorgehen!).
Was diese Männer auch nicht verstehen ist, dass Neuronen zudem jeweils eine leicht unterschiedliche Zusammensetzung aktiver TEs aufweisen. Kein Neuron is genetisch gleich. Fast alle sind ungleich! Dies passt genau zu der von mir 2009 beschriebenen Erwartung variabler, TE-gesteuerter Diversität (du kannst das nachschauen in meinen Vortrag über Prozesse des Lernens auf Youtube). Insofern erklärt mangelnde Kenntnis der modernen Genomarchitektur einen Teil der Fehldeutungen von Neukamm und anderen Evolutionisten, die die Funktionsweise des Genoms in seiner räumlich-dynamischen Komplexität nicht verstehen.
Die Praxis, große strukturelle Umbauten als „ein Mutationsereignis“ zu zählen, ergibt nur dann Sinn, wenn man die Ursache bereits im Vorfeld als Mutation festlegt. Damit bestätigt die Zählmethode lediglich die eigene Vorannahme. Selbst wenn es sich um einzelne Ereignisse handeln würde, müsste man nicht-zufällige Mechanismen einbeziehen – genau jene, die ich in meinen Veröffentlichungen vorgeschlagen habe und die zunehmend durch experimentelle Daten gestützt werden. Für zB FOXP2 habe ich gezeigt, dass man nebst genetischen Wunder auch noch Hypermutationen im regulatorischen Teil akzeptieren muss.
Der Ausschluss hochvariabler Regionen wie Tandem-Repeats oder Low-Complexity-Elemente ist ebenfalls theoriegeleitet. Gerade ihr hoher Variationsgrad stellt die Vorstellung rein punktueller Mutationsprozesse infrage. Aus meiner Sicht handelt es sich dabei um echte, funktionelle Variation, wie ich sie in meinen Büchern und Artikeln beschrieben habe. Das erklärt, warum kein Mensch dem anderen vollkommen gleicht – nicht einmal eineiige Zwillinge. Aus diesem Grund habe ich TEs als „Variation-inducing Genetic Elements“ (ViGEs) bezeichnet.
Was die Genregulation betrifft: Die wirklich funktionell relevanten Unterschiede zwischen Arten liegen neben neue Gene (Orphan genes, miRNAs usw) auf dieser Ebene, nicht in den wenigen Prozent (1,2%) Basensubstitutionen. Entscheidend ist die 4D-Organisation des TE-regulierten Genoms, und diese unterscheidet sich – je nach Messkriterium – in teils bis zu 70 % zwischen Mensch und Schimpanse. Ein Paradigma, das nahezu ausschließlich punktuelle Basenunterschiede gewichtet, wird dieser Realität nicht gerecht; es bleibt ein Relikt des alten, darwinistisch geprägten Verständnisses.“
Quellen und weiterführende Links:
- Yoo D, Rhie A, Hebbar P et al. (2025) Complete sequencing of ape genomes. Nature 641, 401–418. https://doi.org/10.1038/s41586-025-08816-3
- https://scienceandculture.com/tag/chimps-and-critics-series
- https://scienceandculture.com/2025/05/fact-check-new-complete-chimp-genome-shows-14-9-percent-difference-from-human-genome/
- Borger P & Scholl B (2025) Der genetische Unterschied zwischen Mensch und Schimpanse beträgt 15%. Studium Integrale Journal, 32. Jahrgang, Heft 2.